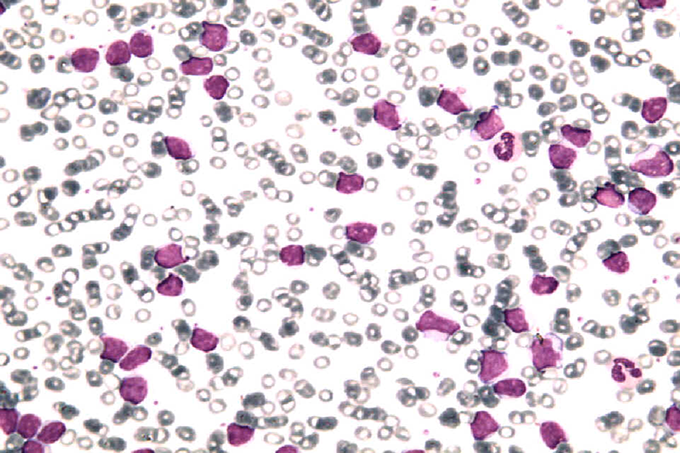

La leucemia linfatica cronica (LLC) è la forma più comune di leucemia nel mondo occidentale; l’età mediana alla diagnosi è circa 70 anni e solo il 15% dei pazienti ha meno di 50 anni. L’incidenza complessiva è di 5 casi su 100.000 persone/anno e aumenta esponenzialmente con l’età. Sebbene la sopravvivenza mediana sia di circa 10 anni, i pazienti con LLC presentano un decorso clinico e una prognosi estremamente variabili: infatti mentre alcuni di loro hanno un decorso indolente e un’aspettativa di vita sovrapponibile a quella di individui sani di pari età, per altri il decorso clinico è aggressivo e la prognosi infausta. La malattia è caratterizzata dalla proliferazione e dal progressivo accumulo di linfociti B nel sangue periferico e negli organi linfoidi (midollo osseo, linfonodi, milza).
Il risultato è l’aumento dei linfociti nel sangue periferico (> 5000/microlitro), l’aumento delle dimensioni dei linfonodi al collo, ascelle e inguine (linfoadenomegalia), l’aumento delle dimensioni della milza (splenomegalia). Per distinguere una linfocitosi da LLC da una linfocitosi di altre malattie linfoproliferative o infettive è fondamentale lo studio morfologico della striscio di sangue periferico e l’immunofenotipo. La morfologia della LLC può essere tipica (80%) o atipica. Alla tipizzazione immunofenotipica i linfociti sono positivi per gli antigeni di superficie CD5, CD19, CD23, debolmente positivi per CD20 e CD22. L’insorgenza della malattia non sembra influenzata dall’esposizione ad agenti chimici o a radiazioni ionizzanti. Invece diversi studi hanno dimostrato la possibilità che fattori genetici possano predisporre a sviluppare la malattia: nel 10% dei pazienti un’indagine familiare rivela la presenza di altri casi di LLC o altre malattie del sistema linfatico.
 Nei 2/3 dei casi all’esordio il paziente è asintomatico e la diagnosi viene posta in seguito al riscontro occasionale di linfocitosi all’esame emocromocitometrico, con- fermata dall’immunofenotipo dei linfociti del sangue periferico. In altri casi alla linfocitosi si associano linfoadenomegalia ed eventualmente splenomegalia. Col progredire della malattia possono comparire sintomi conseguenti alla progressiva insufficienza midollare: pallore cutaneo e stanchezza inusuali legati all’anemia, suscettibilità alle infezioni per la neutropenia (riduzione del numero dei granulociti neutrofili), manifestazioni emorragiche legate alla piastrinopenia. Possono inoltre manifestarsi disordini autoimmuni come anemia emolitica e piastrinopenia autoimmune. I sintomi sistemici, caratterizzati da astenia, febbre, calo ponderale, sudorazione profusa, prurito, dolori osteoarticolari, possono essere rilevati alla diagnosi ma sono più frequenti nelle fasi avanzate di malattia. La LLC ha un decorso clinico estremamente variabile pertanto sono stati proposti fattori pro- gnostici allo scopo di predire il decorso clinico di ogni paziente. Principalmente possono essere distinti in fattori prognostici clinici e biologici.
Nei 2/3 dei casi all’esordio il paziente è asintomatico e la diagnosi viene posta in seguito al riscontro occasionale di linfocitosi all’esame emocromocitometrico, con- fermata dall’immunofenotipo dei linfociti del sangue periferico. In altri casi alla linfocitosi si associano linfoadenomegalia ed eventualmente splenomegalia. Col progredire della malattia possono comparire sintomi conseguenti alla progressiva insufficienza midollare: pallore cutaneo e stanchezza inusuali legati all’anemia, suscettibilità alle infezioni per la neutropenia (riduzione del numero dei granulociti neutrofili), manifestazioni emorragiche legate alla piastrinopenia. Possono inoltre manifestarsi disordini autoimmuni come anemia emolitica e piastrinopenia autoimmune. I sintomi sistemici, caratterizzati da astenia, febbre, calo ponderale, sudorazione profusa, prurito, dolori osteoarticolari, possono essere rilevati alla diagnosi ma sono più frequenti nelle fasi avanzate di malattia. La LLC ha un decorso clinico estremamente variabile pertanto sono stati proposti fattori pro- gnostici allo scopo di predire il decorso clinico di ogni paziente. Principalmente possono essere distinti in fattori prognostici clinici e biologici.
Fattori prognostici clinici e stadiazione clinica; vengono utilizzati due sistemi nella pratica clinica, quello di RAI e quello di Binet. La stadiazione, secondo RAI, prevede la suddivisione in 5 stadi correlati ad una diversa sopravvivenza ovvero:
stadio 0 – linfocitosi isolata, stadio I – linfocitosi e adenomegalie, stadio II – linfocitosi ed epato/slenomegalie con o senza adenomegalie, stadio III – linfocitosi e anemia, stadio IV – linfocitosi+piastrinopenia. La sopravvivenza varia da 19 mesi per l’alto rischio (stadio III e IV) a 150 mesi per il basso rischio (stadio 0):
-tempo di raddoppiamento dei linfociti: se inferiore a 6 mesi indica rapida progressione di malattia.
-infiltrazione midollare di tipo diffuso Fattori prognostici biologici.
-stato mutazionale dei geni IGVH. Il 50% dei pazienti con LLC è IGVH non mutato, condizione associata a malattia più estesa e a prognosi più sfavorevole rispetto ai pazienti con IGVH mutato. La mediana di sopravvivenza dei due gruppi è rispettivamente 8 e 25 anni.
-anomalie citogenetiche all’indagine FISH: la delezione del braccio lungo del cromosoma 13 è presente nel 50% dei casi e, se isolata, è favorevole. La trisomia del cromosoma 12 è presente nel 25% dei casi. La delezione del braccio lungo del cromosoma 11 è presente nel 10-20 % dei pazienti e si associa a marcata linfoadenomegalia e sopravvivenza media di 79 mesi. La delezione del braccio corto del cromosoma 17 (8% all’esordio) è il principale fattore prognostico e si associa a progressione di malattia, ridotta sopravvivenza (32 mesi) e resistenza ai farmaci antitumorali.
-mutazioni geniche. TP 53 (5%) si associa a prognosi sfavorevole e chemioresistenza: PFS 23 mesi e OS 29 mesi. NOTCH 1 (10%) si associa a prognosi sfavorevole. SF3B1 (5%) si associa a refrattarietà alle terapie e a ridotta sopravvivenza. BIRC 3 (4%) associato a prognosi sfavorevole. Quando iniziare il trattamento? Ci si basa sui criteri stabiliti dal NCI-WG.
-Malattia asintomatica in stadio iniziale (RAI 0): semplice osservazione con controlli clinici periodici.
-Malattia con rischio intermedio (RAI I- II): si fa terapia solo in presenza di segni clinici o di malattia progressiva.
-Malattia avanzata (RAI III- IV): il paziente va trattato. I criteri di malattia attiva o progressiva sono: comparsa di anemia e/o piastrinopenia, splenomegalia > 6cm dall’arco costale, masse linfoidi > 10cm, aumento della linfocitosi > 50% in 2 mesi o raddoppiamento in 6 mesi, anemia o piastrinopenia autoimmuni non sensibili alla terapia steroidea, presenza di sintomi sistemici.
L’approccio terapeutico alla LLC si è profondamente modificato. Le cure oggi tendono a un controllo molto più pro- fondo della malattia poiché sappiamo che la qualità della risposta è importante per la sopravvivenza libera da malattia.
Oggi, a differenza di 10-15 anni fa, abbiamo a disposizione molti farmaci attivi, alcuni dei quali innovativi ma anche costosi. Quindi di fronte a un paziente con LLC l’ematologo deve decidere se trattarlo, quando trattarlo e come trattarlo. Esistono ovviamente delle Linee Guida e dei criteri basati sull’età del paziente e sulla presenza di patologie associate più o meno rilevanti. Un programma terapeutico intensivo tollerato da un paziente giovane potrebbe comportare effetti collaterali inaccettabili in un paziente anziano e/o con patologie concomitanti. La scelta terapeutica è inoltre condizionata dai parametri diagnostici che andrebbero valutati prima di ogni trattamento in quanto possono modificarsi col progredire della malattia. Un concetto fondamentale è quello di trattare il paziente più tardi possibile, convincerlo a convivere con la malattia e poiché si tratta di pazienti anziani molti di essi non inizieranno mai un trattamento e non moriranno di leucemia. Oggi sappiamo che la terapia può ridurre la taglia di malattia senza eradicarla, ma può esercitare una selezione negativa sui cloni più maligni e resistenti ai farmaci favorendone l’emergenza. Quindi alla ricaduta ci potremmo trovare di fronte a una malattia biologicamente e clinicamente diversa, più aggressiva e resistente, difficile da curare.
I farmaci utilizzati in prima linea sono il Clorambucile, la Fludarabina, la Ciclofo- sfamide, la Bendamustina usati da soli o in combinazione o associati ad anticorpi monoclonali (immunochemioterapia) come il Rituximab, l’Obinotuzumab, l’Ofatumumab e l’Alentuzumab. La combinazione di farmaci che dà i migliori risultati in termini di risposta è l’FCR (Fludarabina, Ciclofosfamide, Rituximab) da utilizzare nei pazienti di età inferiore a 65-70 anni in assenza di comorbidità. Bendamustina e Rituximab rappresentano un’alternativa meno tossica alla FCR anche se un po’ meno efficace in termini di risposta. Con FCR 90% di risposte globali delle quali il 44% sono complete. Nonostante i miglioramenti ottenuti con la terapia di I° linea la LLC rimane una malattia incurabile, caratterizzata da ricadute più o meno frequenti. Nella scelta della terapia di II° linea occorre considerare il tempo trascorso dalla I° terapia, lo schema terapeutico utilizzato, la presenza o meno della delezione del 17p, il performance status e le comorbidità del paziente. La malattia refrattaria (che non ha risposto alla prima linea o che è ricaduta entro breve tempo) è sicuramente più difficile da gestire. Il trapianto allogenico potrebbe rappresentare un’opzione ma data l’età avanzata dei pazienti solo raramente è praticabile. Inoltre la mortalità peritrapiantologica è ancora elevata (25-35%). Le nuove modalità con condizionamento ridotto consentono di proporre il trapianto non mieloablativo in una fascia più ampia di età e di ridurre la tossicità.
Le novità terapeutiche sono rappresentate da farmaci non classicamente chemioterapici che agiscono su bersagli diversi dal DNA cellulare. Tra questi ricordo l’Ibrutinib, l’ABT263 (anti BCL2), la Lenalidomide, l’Idelalisib e il Duvelisib. Questi farmaci non ancora tutti disponibili in commercio, hanno il vantaggio di essere somministrati per via orale, risultano attivi anche nei pazienti resistenti ai chemioterapici e con caratteristiche biologiche sfavorevoli (17p, TP53), sono ben tollerati anche da pazienti anziani non fit. Le risposte complete sono però molto rare per cui sono in corso studi in cui queste nuove molecole vengono associate a chemioterapici e anticorpi monoclonali. Non dimentichiamo però che in un paziente anziano non fit l’obiettivo sarà non la remissione completa ma il controllo di sintomi e il miglioramento della qualità della vita. Tanti progressi in campo biologico e terapeutico hanno trasformato questa malattia da cenerentola a regina.
La sfida che ci attende è saper utilizzare queste conoscenze e questi nuovi farmaci nel paziente giusto, al momento giusto e nel modo giusto.
Pier Paolo Fattori