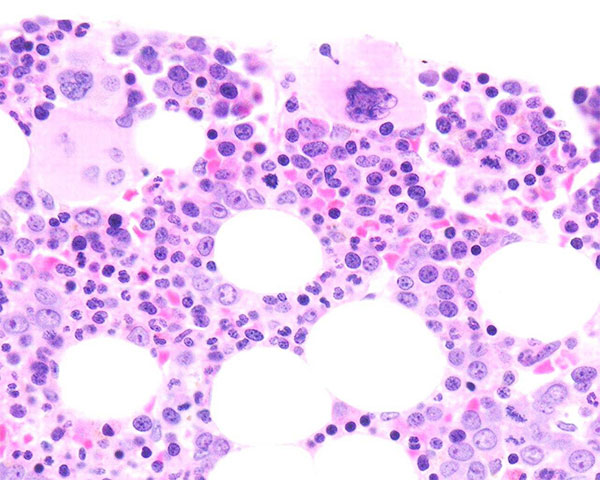

Le Sindromi Mielodisplastiche (SMD) sono un gruppo eterogeneo di patologie ematologiche clonali del midollo osseo che interessano la cellula staminale emopoietica ovvero la cellula che dà origine a tutti gli elementi corpuscolati del sangue: globuli rossi, globuli bianchi e piastrine. Sono caratterizzate da una alterata produzione di cellule ematiche (emopoiesi inefficace) con progressiva insufficienza midollare, alterazioni morfologiche (dette alterazioni displastiche) e cromosomiche (dette alterazioni citogenetiche) a carico delle principali filiere emopoietiche; ciò si traduce in un aumentato rischio di progressione in Leucemia Acuta Mieloide (LAM) rispetto alla popolazione generale. Hanno un’incidenza di circa 4 nuovi casi per 100.000 abitanti per anno e una prevalenza di 7 casi per 100.000 abitanti. Sono tipicamente una patologia dell’età avanzata, l’età mediana si pone fra la sesta e la settima decade di vita. Dal punto di vista clinico le SMD si manifestano con citopenie periferiche (cioè un ridotto numero di cellule ematiche nel sangue periferico): anemia, leucopenia e piastrinopenia variamente associate. Inoltre l’alterata maturazione e le alterazioni citogenetiche comportano anche difetti funzionali di globuli rossi, globuli bianchi e piastrine che, uniti al loro ridotto numero, si riflettono nelle classiche complicanze delle SMD rispettivamente: segni e sintomi legati all’anemia (pallore, affaticabilità, tachicardia, cardiopalmo), infezioni ricorrenti ed emorragie.
La sopravvivenza dei pazienti affetti da SMD varia da alcuni mesi a diversi anni per cui risulta di primaria importanza avere a disposizione sistemi classificativi e prognostici che permettano di predire la probabilità di sopravvivenza e il rischio di evoluzione in LAM allo scopo di definire il più corretto approccio terapeutico. Diversi sistemi classificativi e prognostici sono stati elaborati allo scopo di meglio rispondere a queste esigenze. Nel 1997 è stato proposto un sistema prognostico a punteggio (International Prognostic Scoring System-IPSS; Greenberg PL et al. 1997) che si basa sulla valutazione di 3 parametri: il numero di citopenie all’esame emocromocitometrico, la percentuale di blasti (cellule immature) midollari e la presenza di alterazioni citogenetiche, che permette di identificare 4 diversi gruppi prognostici: a basso rischio (Low), a rischio intermedio 1 e 2 (Intermediate-1, Intemediate-2) e ad alto rischio (High) con sopravvivenza progressi- vamente ridotta e rischio di evoluzione in LAM progressivamente aumentato. Tale sistema prognostico è stato successivamente revisionato nel 2012 (R-IPSS, Greenberg PL et al 2012).
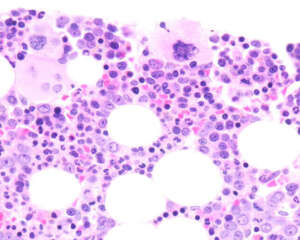 Nel 2012 il gruppo dell’Ematologia di Pavia (Malcovati L. et al) ha introdotto un nuovo sistema prognostico “dinamico” (WHO classification based prognostic scoring system WPSS) che si basa su 3 parametri: la classificazione WHO delle SMD (World Health Organization), l’analisi citogenetica e la richiesta di supporto trasfusionale. È proprio quest’ultimo parametro che rende lo score “dinamico” permettendo, in ogni momento durante il decorso della malattia, di classificare i pazienti in 5 distinti gruppi di rischio (Very Low, Low, Intermediate, High, Very High). Tale sistema permette di predire in modo accurato, la probabilità di sopravvivenza e di progressione in LAM. L’approccio medico e l’atteggiamento terapeutico nei confronti dei pazienti affetti da SMD è molto cambiato negli ultimi anni, grazie a farmaci che hanno dimostrato efficacia e che riescono a modificare la storia naturale della malattia, ma anche grazie a un notevole miglioramento della terapia di supporto. Per molti decenni la terapia delle SMD non ha potuto essere altro che una terapia di supporto: terapia antibiotica e antifungina per le infezioni correlate alla leucopenia (in particolare la neutropenia), trasfusioni di concentrati piastrinici ed eritrocitari per gestire, rispettivamente, la piastrinopenia e l’anemia. Inoltre, durante il decorso della malattia, i pazienti affetti da SMD sono per la maggior parte costretti a ricorrere a emotrasfusioni a causa dell’anemia sintomatica. Per questo motivo, il paziente è a rischio di sviluppare, a lungo andare, un sovraccarico di ferro nell’organismo (Emocromatosi Secondaria). Il danno d’organo (in particolare a carico di fegato e cuore) si aggrava e rende più complesso il quadro clinico dei pazienti. Proprio per evitare l’emocromatosi secondaria, si impone la terapia “ferro chelante” (capace cioè di “legare” il ferro in eccesso e favorirne l’eliminazione attraverso gli organi escretori) specie in quei pazienti che hanno un elevato fabbisogno trasfusionale, elevati livelli di ferritina sierica e una più lunga aspettativa di vita. Per anni è stato disponibile un solo farmaco utilizzabile per questo scopo nelle SMD, la deferoxamina, composto di non semplice gestione e spesso male accettato dai pazienti per l’impatto sulla qualità della vita (necessità di prolungate somministrazioni sottocute mediante dispositivi portatili dedicati). Novità assai importante in questo senso è la disponibilità da alcuni anni di un nuovo chelante orale: il Deferasirox. Il vantaggio di tale farmaco consiste nella somministrazione orale, nell’emivita che consente la monosomministrazione giornaliera e nei pochi e reversibili effetti collaterali causati (soprattutto tossicità gastrointestinale e renale). Studi clinici hanno dimostrato l’efficacia del farmaco nel ridurre sia i livelli di ferritina sierica che i livelli di ferro epatico. Inoltre alcuni pazienti con SMD hanno mostrato un miglioramento dell’ eritropoiesi in seguito a terapia ferro chelante con riduzione del fabbisogno trasfusionale. Secondo le più recenti linee guida la scelta terapeutica, in un paziente con SMD, viene valutata in funzione delle caratteristiche del paziente (età, performance status, comorbidità) e della malattia (score prognostici IPSS-WPSS). Il trattamento comunque è raccomandato per i pazienti che presentino citopenia sintomatica.
Nel 2012 il gruppo dell’Ematologia di Pavia (Malcovati L. et al) ha introdotto un nuovo sistema prognostico “dinamico” (WHO classification based prognostic scoring system WPSS) che si basa su 3 parametri: la classificazione WHO delle SMD (World Health Organization), l’analisi citogenetica e la richiesta di supporto trasfusionale. È proprio quest’ultimo parametro che rende lo score “dinamico” permettendo, in ogni momento durante il decorso della malattia, di classificare i pazienti in 5 distinti gruppi di rischio (Very Low, Low, Intermediate, High, Very High). Tale sistema permette di predire in modo accurato, la probabilità di sopravvivenza e di progressione in LAM. L’approccio medico e l’atteggiamento terapeutico nei confronti dei pazienti affetti da SMD è molto cambiato negli ultimi anni, grazie a farmaci che hanno dimostrato efficacia e che riescono a modificare la storia naturale della malattia, ma anche grazie a un notevole miglioramento della terapia di supporto. Per molti decenni la terapia delle SMD non ha potuto essere altro che una terapia di supporto: terapia antibiotica e antifungina per le infezioni correlate alla leucopenia (in particolare la neutropenia), trasfusioni di concentrati piastrinici ed eritrocitari per gestire, rispettivamente, la piastrinopenia e l’anemia. Inoltre, durante il decorso della malattia, i pazienti affetti da SMD sono per la maggior parte costretti a ricorrere a emotrasfusioni a causa dell’anemia sintomatica. Per questo motivo, il paziente è a rischio di sviluppare, a lungo andare, un sovraccarico di ferro nell’organismo (Emocromatosi Secondaria). Il danno d’organo (in particolare a carico di fegato e cuore) si aggrava e rende più complesso il quadro clinico dei pazienti. Proprio per evitare l’emocromatosi secondaria, si impone la terapia “ferro chelante” (capace cioè di “legare” il ferro in eccesso e favorirne l’eliminazione attraverso gli organi escretori) specie in quei pazienti che hanno un elevato fabbisogno trasfusionale, elevati livelli di ferritina sierica e una più lunga aspettativa di vita. Per anni è stato disponibile un solo farmaco utilizzabile per questo scopo nelle SMD, la deferoxamina, composto di non semplice gestione e spesso male accettato dai pazienti per l’impatto sulla qualità della vita (necessità di prolungate somministrazioni sottocute mediante dispositivi portatili dedicati). Novità assai importante in questo senso è la disponibilità da alcuni anni di un nuovo chelante orale: il Deferasirox. Il vantaggio di tale farmaco consiste nella somministrazione orale, nell’emivita che consente la monosomministrazione giornaliera e nei pochi e reversibili effetti collaterali causati (soprattutto tossicità gastrointestinale e renale). Studi clinici hanno dimostrato l’efficacia del farmaco nel ridurre sia i livelli di ferritina sierica che i livelli di ferro epatico. Inoltre alcuni pazienti con SMD hanno mostrato un miglioramento dell’ eritropoiesi in seguito a terapia ferro chelante con riduzione del fabbisogno trasfusionale. Secondo le più recenti linee guida la scelta terapeutica, in un paziente con SMD, viene valutata in funzione delle caratteristiche del paziente (età, performance status, comorbidità) e della malattia (score prognostici IPSS-WPSS). Il trattamento comunque è raccomandato per i pazienti che presentino citopenia sintomatica.
 In sintesi, i pazienti a basso rischio sono generalmente candidati a trattamenti di supporto per la correzione della citopenia periferica (EPO, G-CSF, trasfusioni). Viceversa, i pazienti a più alto rischio sono candidati a un trattamento specifico, differentemente modulato in base all’età (sono considerati trattabili con terapia specifica pazienti con età <75 anni) e alle condizioni generali. Negli ultimi tempi si sta assistendo a un cambiamento radicale nei confronti di queste patologie, diverse ad oggi sono le opzioni terapeutiche. Le novità più rilevanti riguardano l’uso dei farmaci ipometilanti e la lenalidomide. Sebbene l’uso di farmaci ipometilanti si sia dimostrato efficace nel migliorare la sopravvivenza, il trapianto allogenico, nei pazienti giovani con SMD ad alto rischio, si configura ancora oggi come l’unica terapia in grado di modificare in maniera sostanziale la storia naturale della malattia. Tra gli agenti ipometilanti il trattamento con 5-azacitidina (antimetabolita, somministrato per via sottocutanea) ha mostrato dei risultati promettenti determinando una riduzione del fabbisogno trasfusionale, dell’incidenza di infezioni gravi e di eventi che richiedono ospedalizzazione. Inoltre si è osservato in differenti studi un significativo prolungamento della sopravvivenza e una ridotta incidenza di evoluzione in LAM rispetto all’utilizzo della terapia convenzionale. Sono candidati a terapia con farmaci ipometilanti: pazienti a rischio INT 2/ALTO secondo IPSS, non candidati a trapianto o a chemioterapia. Particolare attenzione deve essere rivolta alla SMD associata a delezione del braccio lungo del cromosoma 5, definita Sindrome 5q-, descritta per la prima volta nel 1974 che rappresenta il primo disordine ematologico associato ad una specifica delezione cromosomica. La terapia dei pazienti affetti da sindrome 5q- e anemia sintomatica trasfusione dipendente è radicalmente cambiata in seguito all’introduzione della lenalidomide. La lenalidomide è un farmaco somministrato per via orale con una attività antineoplastica e potente attività immunomodulatoria in grado di indurre la remissione citogenetica e di eliminare la dipendenza trasfusionale. Principale effetto collaterale associato all’uso della lenalidomide è rappresentato dalla tossicità ematologica (soprattutto neutropenia e piastrinopenia) che richiede successiva modulazione del dosaggio. La diagnostica e la terapia delle SMD hanno attraversato recentemente importanti cambiamenti. Una patologia grave e considerata incurabile fino a pochi anni fa è divenuta una patologia cronica. Non solo i risultati clinici positivi in termini di sopravvivenza e miglioramento ematologico, ma anche di qualità di vita fanno sì che gli ematologi, ma anche le aziende farmaceutiche moltiplichino gli sforzi per ottenere ulteriori miglioramenti nella terapia. I farmaci discussi hanno sicuramente un avvenire, ma è assai probabile che verranno impiegati in combinazione e/o affiancati da altre molecole sviluppate con il preciso obiettivo delle Sindromi Mielodisplastiche.
In sintesi, i pazienti a basso rischio sono generalmente candidati a trattamenti di supporto per la correzione della citopenia periferica (EPO, G-CSF, trasfusioni). Viceversa, i pazienti a più alto rischio sono candidati a un trattamento specifico, differentemente modulato in base all’età (sono considerati trattabili con terapia specifica pazienti con età <75 anni) e alle condizioni generali. Negli ultimi tempi si sta assistendo a un cambiamento radicale nei confronti di queste patologie, diverse ad oggi sono le opzioni terapeutiche. Le novità più rilevanti riguardano l’uso dei farmaci ipometilanti e la lenalidomide. Sebbene l’uso di farmaci ipometilanti si sia dimostrato efficace nel migliorare la sopravvivenza, il trapianto allogenico, nei pazienti giovani con SMD ad alto rischio, si configura ancora oggi come l’unica terapia in grado di modificare in maniera sostanziale la storia naturale della malattia. Tra gli agenti ipometilanti il trattamento con 5-azacitidina (antimetabolita, somministrato per via sottocutanea) ha mostrato dei risultati promettenti determinando una riduzione del fabbisogno trasfusionale, dell’incidenza di infezioni gravi e di eventi che richiedono ospedalizzazione. Inoltre si è osservato in differenti studi un significativo prolungamento della sopravvivenza e una ridotta incidenza di evoluzione in LAM rispetto all’utilizzo della terapia convenzionale. Sono candidati a terapia con farmaci ipometilanti: pazienti a rischio INT 2/ALTO secondo IPSS, non candidati a trapianto o a chemioterapia. Particolare attenzione deve essere rivolta alla SMD associata a delezione del braccio lungo del cromosoma 5, definita Sindrome 5q-, descritta per la prima volta nel 1974 che rappresenta il primo disordine ematologico associato ad una specifica delezione cromosomica. La terapia dei pazienti affetti da sindrome 5q- e anemia sintomatica trasfusione dipendente è radicalmente cambiata in seguito all’introduzione della lenalidomide. La lenalidomide è un farmaco somministrato per via orale con una attività antineoplastica e potente attività immunomodulatoria in grado di indurre la remissione citogenetica e di eliminare la dipendenza trasfusionale. Principale effetto collaterale associato all’uso della lenalidomide è rappresentato dalla tossicità ematologica (soprattutto neutropenia e piastrinopenia) che richiede successiva modulazione del dosaggio. La diagnostica e la terapia delle SMD hanno attraversato recentemente importanti cambiamenti. Una patologia grave e considerata incurabile fino a pochi anni fa è divenuta una patologia cronica. Non solo i risultati clinici positivi in termini di sopravvivenza e miglioramento ematologico, ma anche di qualità di vita fanno sì che gli ematologi, ma anche le aziende farmaceutiche moltiplichino gli sforzi per ottenere ulteriori miglioramenti nella terapia. I farmaci discussi hanno sicuramente un avvenire, ma è assai probabile che verranno impiegati in combinazione e/o affiancati da altre molecole sviluppate con il preciso obiettivo delle Sindromi Mielodisplastiche.
Marianna Norata